Спадкове захворювання обміну речовин Фенілкетонурія (ПКУ) зустрічається рідко, але у випадку захворювання дитини, воно вимагає послідовного харчування з першої хвилини, щоб не допустити пошкодження розвитку мозку та ускладнень, які з ним виникають.
Що таке фенілкетонурія?
.jpg)
© bacsica - stock.adobe.com
Фенілкетонурія це спадкове метаболічне захворювання, при якому в організмі накопичується певний білковий компонент, який насамперед обмежує розвиток мозку у дітей.
У ФРН статистично постраждала лише одна дитина з 10000 народжених. Якщо у цієї дитини діагностовано захворювання рано, фенілкетонурію можна лікувати, і дитина може розвиватися повністю нормально.
причини
Причиною Фенілкетонурія є генетичним дефектом. Це означає, що білковий компонент фенілаланін, який міститься у всіх продуктах, що містять білки, і, отже, приймається разом з їжею, вже не може бути розбитий. У здорових людей з цією метою діє фермент, який у людей з фенілкетонурією, залежно від тяжкості, лише частково працює або повністю припинив свою роботу.
Симптоми, недуги та ознаки
Якщо стан не діагностується і не лікується відразу після народження, перші ознаки проявляються приблизно в тримісячному віці. Дозрівання мозку порушено, мозок не росте адекватно, що видно у дітей старшого віку значно меншою головою. Психічний розвиток відстає. Коли вони досягають статевої зрілості, вони мають серйозні порушення інтелекту.
Через пошкодження клітин мозку діти страждають від підвищеної збудливості, що часто викликає епілептичні припадки. М’язові клітини також уражені хворобою. М'язи напружуються, як спазми (спастичність), що ускладнює нормальні рухи. Крім того, трапляються розлади поведінки у вигляді гіперактивності та агресивності. Діти схильні до неконтрольованих спалахів гніву.
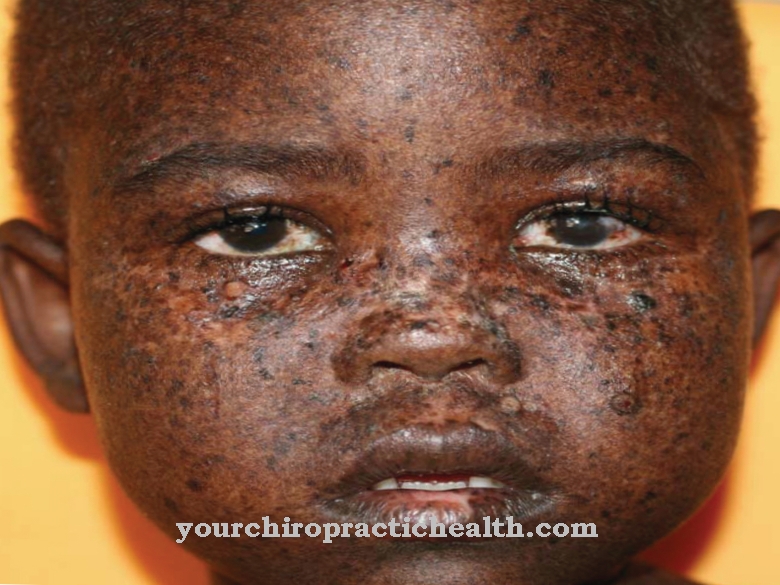
Типовою ознакою ПКУ є запах ацетону, який видають постраждалі. Він обумовлений утворенням в організмі фенілацетату. Ця речовина виводиться через потові залози, а також міститься в сечі. Характерна поява страждаючих ПКУ.
Оскільки їх оранізм не може достатньо продукувати пігмент меланін, у них часто є біло-русяве волосся, дуже світла, чутлива шкіра і світло-блакитні, прозоро мерехтливі очі. Розлад пігментації може спричинити екземоподібні висипання на шкірі. Виразність симптомів залежить від того, наскільки сильно обмежений метаболізм і відхиляється від норми.
Діагностика та перебіг
The Фенілкетонурія можна визначити за допомогою аналізу крові, який зазвичай проводять при скринінгу новонароджених у рамках так званого профілактичного обстеження U2. Проведений тут аналіз крові визначає, серед іншого, рівень фенілаланіну немовляти.
Оскільки метаболізм порушений при захворюванні фенілкетонурією і фенілаланін більше не може бути порушений, він все більше осідає в крові і може бути виявлений тут. Підвищене значення фенілаланіну від 1 до 2 міліграм на децилітр в крові вказує на захворювання з фенілкетонурією.
Якщо підозра на фенілкетонурію під час вагітності, тест навколоплідних вод може бути використаний для проведення внутрішньоутробного ДНК-тесту на немовляти та постановки діагнозу. Якщо діагностована фенілкетонурія, будуть проведені додаткові аналізи для визначення ступеня тяжкості стану, які будуть використані для цільового лікування.
Якщо фенілкетонурію не лікувати, перші симптоми метаболічного захворювання з’являються приблизно через три місяці, які швидко призводять до серйозних ускладнень, включаючи затримку психічного розвитку до важких порушень інтелекту, порушене напруження м’язів і навіть епілептичні припадки.
Ускладнення
У будь-якому випадку фенілкетонурію потрібно лікувати відразу після народження дитини. Якщо таке лікування не відбудеться, це може призвести до серйозних скарг на ріст і розвиток дитини, які не можна компенсувати згодом. Постраждалі зазвичай страждають порушенням обміну речовин через фенілкетонурію.
Це також призводить до підвищеної дратівливості, так що діти здаються агресивними. Затримка рухового та психічного розвитку також може стати очевидною і значно знизити якість життя постраждалої людини. У багатьох випадках батьки також страждають від депресії або інших психологічних порушень. Постраждалі також проявляють неприємний запах тіла, так що уражені діти та підлітки можуть стати жертвами дражнити або знущатися.
Крім того, виникають епілептичні припадки, які в гіршому випадку можуть призвести до смерті. Пігментні порушення можуть спостерігатися і у дитини. Лікування фенілкетонурії не пов'язане з ускладненнями. Правильне харчування може запобігти небажаним подіям, щоб не було більше ускладнень у дорослому віці.
Коли потрібно звертатися до лікаря?
Оскільки фенілкетонурія може призвести до серйозного пошкодження мозку дитини, в даному випадку цю хворобу потрібно негайно лікувати. Рання діагностика дуже важлива для запобігання подальших ускладнень або порушення розвитку дитини. Як правило, фенілкетонурія стає помітною через уповільнення розвитку дитини, також відіграють роль інтелектуальні вади та епілептичні припадки.
У разі виникнення епілептичного нападу негайно викликайте лікаря швидкої допомоги або негайно вирушайте до лікарні, оскільки це єдиний спосіб уникнути подальшого пошкодження. У молодому віці фенілкетонурія часто призводить до спалахів гніву або тяжких порушень поведінки. Якщо ці симптоми також виникають, необхідний візит до лікаря. Пігментні порушення або плями на шкірі також можуть свідчити про це захворювання.
Лікування фенілкетонурії залежить від точних симптомів і проводиться різними фахівцями. Часто корисне також психологічне лікування, в якому також можуть брати участь батьки чи родичі. При ранньому діагностуванні та лікуванні тривалість життя пацієнта зазвичай не впливає негативно.
Лікування та терапія
Коли а Фенілкетонурія був поставлений діагноз, зазвичай його можна лікувати дуже добре, так що дітям не потрібно страждати від аномального розвитку мозку, а натомість розвиватися повністю нормально психічно.
Однак це можливо лише при послідовному дотриманні дієти з низьким вмістом фенілаланіну. Не існує іншого варіанту терапії, наприклад, на основі лікарських засобів, для лікування фенілкетонурії та уникнення пошкодження мозку. Особливо важливо майже повністю уникати продуктів, що містять фенілаланін у період, коли розвивається мозок - тобто в період від народження до статевого дозрівання.
Чим раніше розпочато дієту, тим краще прогресує фенілкетонурія. З цієї причини дієту зазвичай починають, як тільки народжується новонароджене, і немовлятам дають спеціальну формулу, яка майже не містить фенілаланіну замість грудного молока. Навіть якщо дієта з низьким вмістом фенілаланіну є особливо важливою до статевого дозрівання, лікарі рекомендують довічну дієту з низьким вмістом фенілаланіну, щоб не обмежуватися хворобою до старості.
Однак в раціоні для лікування фенілкетонурії важливо, щоб дієта містила мало фенілаланіну, але не повністю перешкоджала всмоктуванню фенілаланіну. Оскільки фенілаланін - життєво важлива амінокислота, яка особливо необхідна для росту дітей, які страждають на фенілкетонурію. Однак певного значення в крові не слід перевищувати. Тому люди з фенілкетонурією повинні регулярно здавати аналізи крові.
Прогноз та прогноз
Прогноз при класичній фенілкетонурії надзвичайно хороший, якщо діагноз ставиться рано, бажано у новонародженої дитини. Якщо дієта з низьким вмістом фенілаланіну призначається в ранньому грудному віці, не слід очікувати жодних фізичних чи психічних порушень. Постраждалі мають нормальне життя та середню тривалість життя.
Важливою умовою позитивного прогнозу є суворе дотримання дієти, особливо в перші шість років життя, коли мозок особливо розвивається. Жінки з фенілкетонурією також можуть завагітніти і мати своїх дітей. Хоча це спадкове метаболічне захворювання, шкоди дитині не слід очікувати. Однак дієти потрібно дотримуватися послідовно під час вагітності.
Без дієти з низьким вмістом фенілаланіну захворювання призводить до порушень розвитку мозку в ранньому дитинстві. Неврологічні відхилення можна помітити вже з 4-го місяця життя. Оскільки пошкодження мозку є незворотним, немає можливості вилікувати. Коефіцієнт інтелекту зазвичай буде постійно нижче норми. Крім того, можуть виникати судоми, порушення поведінки та рухові порушення.
Особливим випадком є нетипова форма фенілкетонурії, яка характеризується дефіцитом коензиму ВН4 (тетрагідробіоптерину). Прогноз інтелектуального розвитку у цих пацієнтів неможливо оцінити послідовно. Незважаючи на ранню дієту, може статися пошкодження нервової системи.
профілактика
Оскільки це a Фенілкетонурія Якщо це спадкове метаболічне захворювання, немає ніякого способу запобігти фенілкетонурію. Однак якщо у вас фенілкетонурія, дієта з продуктами з низьким вмістом фенілаланіну може запобігти симптоми та ускладнення, які зазвичай виникають при фенілкетонурії.
Щоб захистити власних дітей від наслідків захворювання фенілкетонурією, люди, які вже вагітні, партнер яких вагітна або які планують вагітність, повинні суворо дотримуватися дієти з низьким вмістом фенілаланіну. Якщо дитина захворіє, слід звернути увагу на дієту з низьким вмістом фенілаланіну для благополуччя дитини.
Догляд за ними
Оскільки фенільна кентонурія є одним із спадкових порушень обміну речовин і не може бути вилікувана, а лише лікуватися, то після догляду та лікування значною мірою однакові. Це насамперед включає дієту з низьким вмістом фенілаланіну та регулярний контроль рівня ПА в крові. Ці заходи повинні суворо дотримуватися до шестирічного віку, але необхідні для життя.
При атиповій фенілкетонурії коензим BH4 також повинен бути доповнений. Якщо лікування було розпочато занадто пізно, подальший догляд також може поширюватися на наявні ушкодження. Вони здебільшого знаходяться в області розвитку мозку і можуть різнитися за ступенем вираженості.
Може бути корисним розпочати заходи підтримки (наприклад, трудотерапію) на ранній стадії, щоб протидіяти поведінковим розладам. До них відносяться проблеми зі сном, необхідність контролю, самопошкодження чи антисоціальна поведінка, тяжкість яких часто може бути успішно пом’якшена. У разі рухових розладів цільова фізіотерапія може покращити симптоми.
Якщо дотримуватися призначених заходів, сьогодні багато пацієнтів можуть вести майже нормальне життя. Коли ви досягаєте статевої зрілості, найчутливіша фаза закінчується. Більш великих, неврологічних дефектів зазвичай більше не можна очікувати, навіть якщо коливання рівня ПА в рази можуть погіршувати хімію мозку. Прийом дофамінових препаратів допомагає проти цього.
Ви можете зробити це самостійно
При класичній фенілкетонурії на першому плані дотримується вегетаріанської дієти без аспартаму з джерелом фенілаланіну. Новонароджених годують виключно формулою без фенілаланіну. Фруктові та овочеві келихи підходять як додаткова їжа. Батьки дітей, які страждають на ПКУ, повинні обчислити вміст фенілаланіну у всій їжі, яку вони годують, і послідовно дотримуватися граничних значень.
Пізніше дитяче харчування буде замінено протеїновим порошком, спеціально розробленим для цього захворювання. Суміш амінокислот для пацієнтів з ПКУ необхідно додавати до кожного прийому їжі. Він утворює довічний основний компонент необхідної дієти з низьким вмістом фенілаланіну. Тому доцільно, щоб малюк звик до запаху та смаку порошку грайливо.
Зараз існує широкий спектр навчальних та кулінарних курсів для постраждалих дітей різного віку. Ці освітні заходи просувають особисту відповідальність поетапно. У той же час вони підтримують позитивне поводження з харчовими обмеженнями. Готові продукти з низьким вмістом фенілаланіну дозволяють молодим пацієнтам брати участь у екскурсіях та шкільних поїздках. Батьки хворих дітей піддаються сильному організаційному та психологічному стресу. Їм корисно обмінятися ідеями з постраждалими в групах самодопомоги.
Дорослі пацієнти з фенілкетонурією, які бажають мати дітей, повинні бути особливо обережними при плануванні своїх сімей: лише дотримання суворої дієти забезпечує нормальний рівень фенілаланіну під час вагітності. Нормальні значення абсолютно необхідні в момент зачаття, щоб уникнути серйозної шкоди для ненародженої дитини.





.jpg)