The Синдром Жубер характеризується вродженою вадою розвитку стовбура мозку, а також агенезом (гальмування пригнічення, відсутність прикріплення, наприклад, церебральні бруски, апендикс). Гіпоплазія (недорозвинення) мозочка черв'яка також може існувати. Пацієнти, які страждають від цього аутосомно-рецесивного генетичного дефекту, виявляють, серед іншого, аномальну дихальну поведінку та атаксію.
Що таке синдром Жуберта?

© Сашкін - stock.adobe.com
Люди с Синдром Жубер страждають від порушень розвитку центральної нервової системи та виникаючих функціональних порушень. Медичні дослідження суперечливі, чи слід це генетичне порушення кваліфікувати як захворювання саме по собі.
У уражених пацієнтів спостерігається безліч різних симптомів. Через це остаточний діагноз утруднений. JB характеризується великою неоднорідністю локусу гена. Поки було виявлено багаторазові генні мутації. Мутаційний аналіз дуже обширний.
причини
Синдром Жубер належить до групи первинних циліофатій. При цьому генетичному порушенні первинної війки або базального тіла можуть виникати різні типи порушень розвитку. Як спеціальні клітинні процеси, реснички виконують різні завдання. Вони діють як датчики хіміо-, механо- та осмосу та беруть участь у багатьох сигнальних шляхах. Крім того, вони забезпечують нормальний розвиток органів.
Вони підтримують тканинний гомеостаз основних процесів розвитку. Велика кількість залучених білків утворює складну мережу завдяки взаємодії. Якщо крім основних симптомів порушені й інші органи, тоді присутній JSRD (синдром, пов'язаний з синдромом Джоубера). Це вторинне захворювання характеризується подальшими органовими проявами, що стосуються нирок, печінки та очей.
Це генетично гетерогенний синдром. Лікарі виявили вади гена NPHP6 / CEP290 (кодування для нефроцистину-6) або гена NPHP8 / RPGRIP1L (кодування для нефроцистину-8). Інші генні мутації - MKS3, ARL13B, AHI1, CC2DA2, TMEM216 та INPP5E. Лише у кількох пацієнтів є мутації в NPHP4 та NPHP1.
Симптоми, недуги та ознаки
Патогномонічна особливість - це «молярний зубний знак» (МТС), який можна визначити, використовуючи «осьову магнітно-резонансну томографію головного мозку Т1». Ця особливість характеризується агенезією або гіпоплазією мозочка черв'яка або мозочка. Крім того, сильно втягується задня інтерпендикулярна ямка (яма між ніжками головного мозку), а мозочкові стебла мають чітко виражену верхню форму через деформацію середнього мозку.
Крім МТС, пацієнти часто страждають від респіраторних розладів, атаксії, м’язової гіпотензії та психомоторної відсталості. 8–19 відсотків постраждалих демонструють післяоксіальну полідактилію (кілька пальців), а шість відсотків потиличну (менінго) енцефалоцеле, в якій задня частина мозку випинається.
Ця деформація вперше була зафіксована в 1969 році. Поширеність становить приблизно 1: 100 000, співвідношення, яке показує, як рідко з’являється захворювання. Зафіксовано лише сто випадків з моменту першого медичного обстеження. Оскільки цей генетичний дефект зустрічається в різних формах і варіантах, лікарі припускають багаторазові зміни генетики.
Точна аномалія ще не остаточно підтверджена. Однак мутація Х-хромосоми вважається певною. Цей розлад передається на основі аутосомно-рецесивного успадкування. Залучаються відсутня вермішева мозочка (мозочок, мозочок), пошкодження сітківки та помітна райдужна оболонка.
Симптомами та скаргами, які часто зустрічаються в неонатальний період, є ністагм та неправильний характер дихання, як епізодична тахіпноя та апное. У маленьких дітей може розвинутися гіпотонія. З настанням віку розвивається дисбаланс і нерівномірна хода (атаксія). Ці основні симптоми також відомі як рухові віхи.
Пацієнти мають різний рівень когнітивних здібностей і можуть бути сильно порушені, але вони також можуть проявляти нормальний рівень інтелекту. Можлива також окуломоторна апраксія (порушення руху).
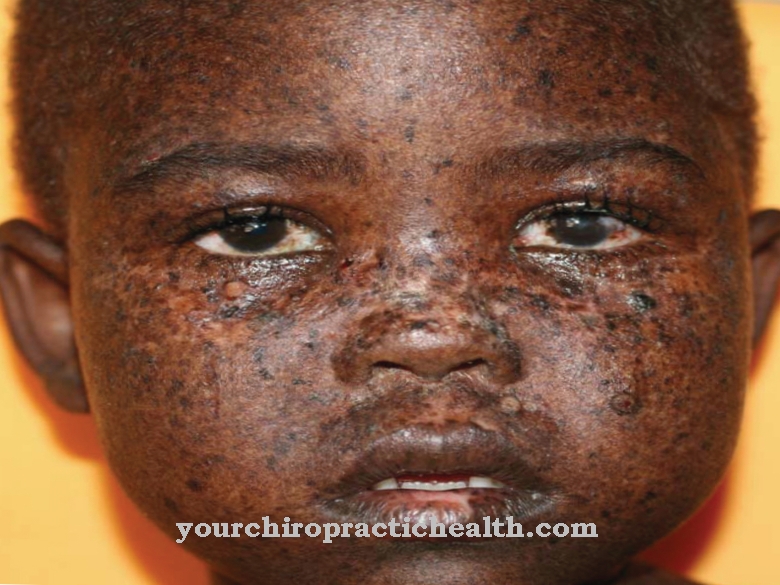
Характерними для цього генетичного дефекту є черепно-лицьові відхилення, такі як велика голова, округлі та високі брови, чіткий (виступаючий) лоб, деформований рот, ритмічно рухається і виступаючий язик і глибоко поставлені вуха. Іноді симптомами є нефрофтиз, дистрофія сітківки та полідактилія.
Діагностика та перебіг захворювання
Діагноз ставлять на підставі цитованих раніше характерних етапів атаксії, гіпотонії, окорухової апраксії, відкритої вербельної мозолі після 18 тижня вагітності та затримки розвитку. Крім того, зроблено характерне нейрорадіологічне знаходження на МРТ, МТС (знак молярного зуба).
Ця особливість, відома як молярний знак, обумовлена вадами деформації пасти і середнього мозку, а також гіпоплазією малого черв'яка. Диференціальні діагнози ставляться на основі захворювань, тісно пов'язаних з JS, такими як JSRD (розлад, пов’язаний з синдромом Джоубера), мальформація Денді-Уолкера (неправильно сформувався мозок мозку без МТС), типи 1 та 2 окорухової апраксії, понто-церебральна гіпоплазія та атрофія, 3-c Синдром, орофаціо-цифрові синдроми II і III, а також синдром Меккеля-Грубера.
Етап I включає "панельний аналіз на основі послідовності наступного покоління" генів JBTS5 (53 кодуючі екзони), JBTS3 (26 кодуючих екзонів), JBTS6 (28 кодуючих екзонів) та JBTS9 (36 кодуючих екзонів). Ген JBTS4 тестують на гомозиготну делецію за допомогою мультиплексної ПЛР. На II стадії інші гени JB аналізуються за допомогою ПЛР (процес, який дублює послідовності генів у ланцюзі ДНК залежно від ферменту) та подальше Сангерне секвенування, залежно від фенотипічних особливостей, що відповідає зменшенню частоти мутації.
Для того, щоб виключити хромосомні дисбаланси, проводять диференційно-діагностичний аналіз масиву SNP. Якщо є родинність або якщо в сім'ї відомо декілька хворих, лікарі проводять скринінг гомозиготності за допомогою куплетного аналізу в мікросупутниковому маркері, що фланкує ген, і подальшому геновому аналізі за допомогою секвенурації Сангера. Два-десять мілілітрів крові ЕДТА відбирають у дітей як діагностичний матеріал, у дорослих кількість становить п'ять-десять мілілітрів.
ДНК або тканинний матеріал також підходить. Стадія I: Матеріал геномної ДНК досліджують на наявність дублікацій або делецій за допомогою кількісного аналізу гена NPHP1 за допомогою MLPA. Дуже невелика кількість ДНК у геномі досліджується на делеції та дублювання окремих екзонів (сегментів генів). II етап: кодовані екзони генів, визначених досі, оцінюються за допомогою частот наступного покоління. Місця сплайсингу збагачені гібридизацією зонда.
Ускладнення
Синдром Жубер призводить до того, що більшість пацієнтів страждають різними недугами. Зазвичай це призводить до низького росту, порушення дихання і, крім того, до затримки. Психічний розвиток дитини також може бути обмежений. Утруднення дихання також може призвести до задишки, яку обов'язково потрібно лікувати.
Не рідкість батьки людини страждають від важкої депресії або інших психологічних порушень. Пацієнти також виявляють порушення рівноваги і часто страждають від обмеженої рухливості. Не рідкість є дискомфорт в очах і вухах, що призводить до зниження слуху або проблем із зором. Якість життя пацієнта значно знижується синдромом Жубер.
За допомогою різних методів терапії можна обмежити і лікувати синдром Жубер. На жаль, причинно-наслідкове лікування не можна проводити. У надзвичайних ситуаціях також можна проводити аварійну вентиляцію, якщо є недолік дихання. Особливих ускладнень у самому лікуванні немає. Загалом, не можна передбачити, чи зменшиться тривалість життя пацієнта за допомогою синдрому Жубер.
Коли потрібно звертатися до лікаря?
Майбутня мама повинна брати участь у всіх доступних обстеженнях під час вагітності. В обстеженнях вивчається стан здоров'я вагітної, а також стан ненародженої дитини. Оскільки синдром Жубер можна діагностувати ще на 18-му тижні вагітності, доцільно використовувати профілактичні медичні огляди, рекомендовані медичними страховими компаніями. Крім того, якщо в історії батьківських предків є генетичний дефект, генетичне консультування та обстеження, як правило, доцільно.
У тому випадку, коли в утробі матері не виявлено порушення, автоматичні огляди акушерами та педіатрами проводяться відразу після пологів. Порушення дихання можуть бути виявлені під час цих обстежень. Якщо батьки дитини помічають незвичні розбіжності, які раніше не виявлені, спостереження слід обговорити з лікарем. Якщо є якісь фізичні особливості, низький ріст або деформації, слід звернутися до лікаря.
Якщо в прямому порівнянні з дітьми того ж віку помічені проблеми з мовою або розумове недорозвинення, слід звернутися до лікаря. Для з’ясування причини необхідні розслідування. Чим раніше буде поставлений діагноз, тим раніше можуть бути розпочаті цільові терапії для підтримки дитини. Отже, консультація з лікарем повинна проходити при перших ознаках порушення.
Лікування та терапія
Батьки мають право на генетичне консультування. Варіанти лікування настільки ж різноманітні, як і причини цього захворювання різноманітні. У випадку порушень рухового розвитку та гіпотензії вступають у програму освітньої підтримки, мови, професійної та трудової терапії, які можуть сприятливо впливати на перебіг захворювання.
Людям, які страждають порушеннями дихання, також можуть бути замінені киснем або вентиляцією. Пацієнти з легкими симптомами мають позитивний прогноз. Сильно постраждалих пацієнтів повинен опікувати експертний довідковий центр.
Прогноз та прогноз
Прогноз при синдромі Джоуберта поганий. Цей синдром є генетичним розладом. Існуючі медичні, наукові та правові вимоги цього неможливо вилікувати. Дослідники та лікарі юридично не дозволяють змінювати генетичні умови людини за допомогою втручань. З цієї причини лікування спрямоване на використання терапії, яка покликана поліпшити існуючу якість життя. Без використання медичної допомоги ще більше знижується самопочуття пацієнта.
Чим раніше синдром можна буде діагностувати і лікувати, тим краще будуть результати. У надзвичайних ситуаціях показана екстрена вентиляція відповідної людини, інакше пацієнт може передчасно померти. Хоча численні терапії поєднуються та застосовуються в індивідуальному плані лікування, наявне захворювання може призвести до вторинних розладів. Вони погіршують загальний прогноз.
Існуючі функціональні порушення або інші обмеження руху можуть призвести до психічних захворювань. У багатьох пацієнтів зафіксовано тимчасову або стійку депресію, перепади настрою або зміни особистості. Це представляє додатковий тягар для відповідної людини та навколишнього середовища. Повсякденне життя хворого на синдром Жубер часто може бути вдається лише за достатньої допомоги та підтримки родичів. Порушення рівноваги та атаксія стають більш важкими з віком.
профілактика
Оскільки точна генетична причинна ситуація ще не була визначена остаточно, в клінічному сенсі немає профілактичних заходів. Єдиний спосіб протидії порокам розвитку в організмі людини - вести здоровий спосіб життя.
Догляд за ними
У більшості випадків у пацієнта з синдромом Жубер немає доступних прямих чи спеціальних варіантів спостереження, тому людина, яка страждає, в першу чергу залежить від швидкої та, перш за все, ранньої діагностики захворювання. Чим раніше захворювання розпізнається, тим краще буде подальший перебіг. Тому бажано звернутися до лікаря при перших симптомах і ознаках.
При цьому захворюванні уражена людина зазвичай залежить від інтенсивної терапії та терапії, яка може полегшити симптоми. Допомога та підтримка батьків та близьких родичів також дуже затребувані для того, щоб постраждала людина могла вести нормальне життя. Часто вправи від фізіотерапії або фізіотерапії також можна виконувати у власному будинку, що може полегшити симптоми.
Симптоми не завжди можуть бути повністю полегшені. Контакт з іншими страждаючими синдромом Жубер також може бути дуже корисним, оскільки не рідкість обмін інформацією. Як правило, тривалість життя ураженої людини цим захворюванням не скорочується.
Ви можете зробити це самостійно
Синдром Джоуберта невиліковний, а також допомогти щоденно. Симптоми вродженого захворювання в більшості випадків неминучі. І все-таки можливо, що деякі з них будуть полегшені.
Оскільки дихання особливо порушено у постраждалих, це є вихідним моментом. Оптимізований клімат приміщення може бути корисним. Сухе нагрівальне повітря може посилити проблеми з диханням. Повітря, який занадто холодний, має той же ефект. В ідеалі температура в приміщенні близько 20 ° C, а вологість повітря близько 50 відсотків. Зокрема, кімнатні рослини можуть сприяти оптимальному клімату в приміщеннях. Як варіант, вологі рушники також можна розмістити в приміщенні, щоб підтримувати вологість на бажаному рівні. Клімат у приміщенні можна відстежити за допомогою гігрометра. Ще одна відправна точка, яка також орієнтована на дихання - дихальні вправи. Регулярне використання покращує сприйняття інакше автоматичного процесу. Таким чином ви можете запобігти занадто швидко дихати і призупинити дихання.
Це також має сенс, якщо постраждалі не сплять одні в кімнаті. Родичі можуть помітити паузи в диханні під час сну і розбудити пацієнта або стимулювати їх до дихання. Але це лише обережність.
.jpg)

.jpg)