Всього існує 45 різних захворювання лізосомального зберігання, які є гетерогенною групою вроджених захворювань обміну речовин. Люди, які страждають від будь-якого з цих захворювань, мають генетичний дефект. Усі хвороби зберігання мають одне спільне: певний фермент або відсутній, або лише частково функціонує.
Що таке захворювання лізосомального зберігання?

© designua - stock.adobe.com
Ці захворювання вродженого зберігання зустрічаються рідко, оскільки страждають менше п'яти на 10000 людей. Різні захворювання мають дуже різний перебіг, і симптоми можуть сильно відрізнятися.
Найвідоміші форми о хвороба лізосомального зберігання - хвороба Фабрі, хвороба Гошера, хвороба Помпе та мукополісахаридоз (MPS). Їх часто називають "сиротами медицини", оскільки шлях до конкретного діагнозу та відповідної терапії може бути дуже довгим. Іноді постраждалі можуть зайняти роки, щоб дізнатися, що з ними відбувається.
причини
Хвороби лізосомального зберігання характеризуються певними формами спадкових захворювань обміну речовин. Пацієнту не вистачає важливого ферменту, який забезпечує безперебійність обміну речовин. У менш вираженій формі цей фермент принаймні не присутній у достатній кількості.
Завдання ферментів - утилізувати забруднювачі та відходи речовин, які накопичуються в організмі людини за допомогою метаболізму через лізосоми, або переробляти їх знову таким чином, щоб симптоми не виникали.
Якщо є дефіцит ферментів, цей плавно функціонуючий цикл утилізації вже не гарантується. Шкідливі речовини осідають у клітинах і порушують метаболічний цикл. У початковій фазі порушення не мають помітного ефекту, є лише кілька обмежень. Однак якщо це порушення обміну речовин не лікується внаслідок дефіциту ферментів, симптоми посилюються через те, що клітини сильно збільшуються.
Симптоми, недуги та ознаки
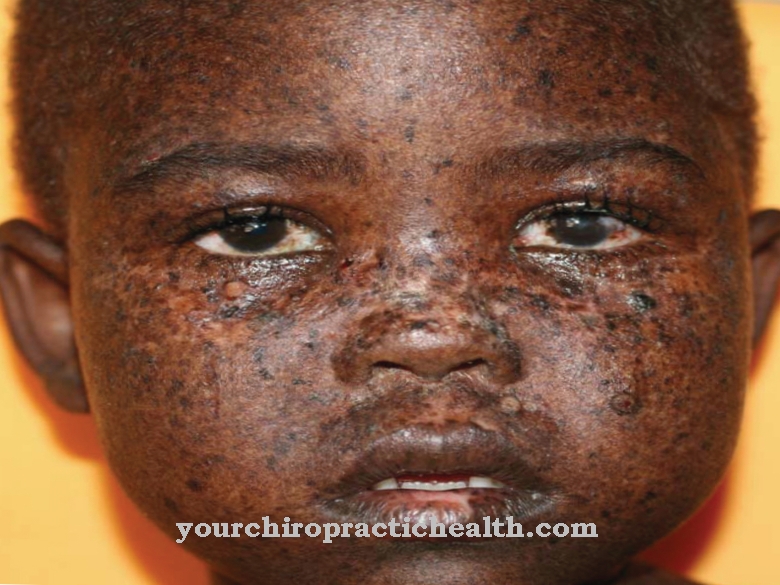
У гіршому випадку вони підпадають. Наслідки - пошкодження кісток, нервової системи, селезінки, нирок, м’язів чи серця. Через знижену або відсутню активність ферментів, хвороба Фабрі призводить до того, що жир (глоботріаосілцерамід, Gb3) зберігається в клітинах. Ці небажані відкладення можуть призвести до сильного болю в пальцях ніг або пальців, інсульту та ураження нирок.
Діагностика та перебіг захворювання
Ця клінічна картина впливає одночасно на різні системи: кровоносні судини, нирки, серце та нервову систему. Автосомно-рецесивна успадкована хвороба Гоше викликає мутацію ферменту "бета-глюкоцереброзидаза" і призводить до накопичення субстрату всередині клітин, особливо в макрофагах (клітини копальника), що належать до ретикуло-ендотеліальної системи. Змінюється аналіз крові, збільшується печінка та селезінка, болять кістки.
Захворювання є прогресуючим і здебільшого етнічним, оскільки зустрічається в більшості випадків у людей єврейського походження. Хвороба Помпе також відома як "дефіцит кислотної мальтази". Клінічна картина належить до групи глікогенезу II типу. У уражених не вистачає ферменту «альфа-1,4-глюкозидаза» (кислотна мальтаза) або він недоступний у достатній кількості. Через порушення руйнування глікогену в м’язах пацієнти страждають від руйнування м’язових клітин у вигляді зберігання цукру.
Мукополісахаридоз I типу (MPS), також відомий як хвороба Хантера, має різні клінічні причини. Хвороба Герлера є найбільш важкою формою, а хвороба Шейє - в кінці клінічного патогенезу. Між цими двома формами прогресування існують різні переходи. Найвизначнішою особливістю є порушення руйнування вуглеводів, що накопичуються в лізосомах клітин.
Хворі на мисливця можуть відчувати низький ріст, збільшену селезінку та печінку, грубі риси, потовщену шкіру, розширений язик та утруднене дихання. Крім того, скелет часто змінюється в області таза, хребта, кісток руки та черепа. Можливі пупкові та [[пахові грижі].
Ускладнення
У більшості випадків симптоми або ускладнення проявляються при цьому захворюванні дуже пізно. Через це він діагностується пізно, так що раннє лікування неможливе в більшості випадків. Без лікування в міру прогресування захворювання виникають різні скарги та пошкодження внутрішніх органів.
Особливо страждають нирки, печінка та селезінка. Серце також може бути уражене цим захворюванням, яке може призвести до серцевої смерті в гіршому випадку. Крім того, відбувається ураження нирок, і ті, хто страждає, часто страждають від болю в пальцях ніг або пальців. Параліч також може виникнути, якщо мозок був пошкоджений цим захворюванням. Печінка та селезінка можуть збільшуватися, а також викликати сильний біль.
Не рідкість кістки ураженої людини також крихкі і болючі. Лікування цього захворювання виявляється важким. У багатьох випадках тривалість життя постраждалої людини значно скорочується. При застосуванні ліків зазвичай не виникає особливих ускладнень. Однак позитивний перебіг захворювання не може бути гарантований у кожному випадку.
Ви можете знайти ліки тут
➔ Ліки від болюКоли потрібно звертатися до лікаря?
Випадання волосся, проблеми із суглобами та порушення органів - можливі ознаки захворювання лізосомального зберігання. Візит до лікаря рекомендується, якщо симптоми повторюються або якщо вони з’являються раптово, не виявляючи причини. Якщо симптоми пов’язані з уже діагностованим дефектом ферменту або іншим серйозним захворюванням, слід звернутися до відповідального лікаря. Нелікована хвороба зберігання може призвести до деменції, безпліддя, невропатій та інших ускладнень, деякі з яких небезпечні для життя. Тому слід обстежити всі можливі симптоми, навіть якщо немає конкретних підозр.
Симптоми захворювання лізосомального зберігання можуть проявлятися поетапно або підступно розвиватися, але завжди потребують обстеження та лікування. Уразливим людям найкраще звернутися безпосередньо до свого сімейного лікаря або до інтерніста. Фактична терапія зазвичай проводиться в спеціалізованій клініці з внутрішніх хвороб, хоча фізіотерапія або психотерапія можуть бути пов'язані залежно від симптомів. Зокрема, показані терапевтичні заходи через часто негативний перебіг захворювання.
Терапія та лікування
Залежно від того, наскільки рано поставлено адекватний діагноз, ці спадкові захворювання можна дуже добре лікувати за допомогою ензимозамінної терапії, так що у людей, які страждають, є набагато менше симптомів і, отже, краща якість життя. Ця замісна терапія застосовується відповідно до клінічної картини.
Людям, які страждають на хворобу Гоше, не вистачає «ферменту ß-глюкоцереброзидази», який виробляється біотехнологічно та вводиться в організм пацієнта. Лізосоми діють ефективно і здатні поглинати речовини з їх найближчого оточення. З цієї причини штучно використовувані ферменти модифікуються таким чином, щоб їх можна було ідеально доставити лізосомам.
Макрофаги (фагоцити) розщеплюють накопичені в клітинах глюкоцереброзиди. Цю терапію можна порівняти з інсулінотерапією при цукровому діабеті, з тією різницею, що це не відсутній гормон, а фермент, який не надходить. Організм регулярно розщеплює всі речовини, включаючи штучний фермент, що постачається.
Через цю регулярну руйнування речовини пацієнтам доводиться регулярно проходити цю інфузійну терапію до кінця життя. Ензимозамісна терапія не діє симптоматично, а безпосередньо бореться з причиною спадкового захворювання. Лікарі називають цю терапію причиною. Принципи терапії повинні застосовуватися при всіх чотирьох вищезгаданих поширених захворюваннях зберігання.
Пацієнтів Помпе також лікують інфузійною терапією. При цьому захворюванні поставляється неіснуючий фермент "кислотна альфа-глюкозидаза" і допомагає розщеплювати глікоген, який накопичився в лізосомах м'язів. У пацієнтів із захворюванням типу "мукополісахаридоз I типу" лізосомальний фермент "альфа-ідуронідаза" відсутній або його немає у достатній кількості. Це одне з найрідкісніших захворювань зберігання, при якому молекули цукру накопичуються в органах і тканинах.
Якщо процес нормальний, фермент розщеплює мукополісахариди. Молекули цукру є довголанцюговими і беруть участь у виробленні опорної та сполучної тканини, наприклад, кісток, шкіри, рідин суглобів та хрящів. Якщо нормальний перебіг деградації порушений через нестачу ферменту, в окремих клітинах накопичуються патологічні глікозаміноглікани (GAG). Майбутні варіанти терапії спрямовані на прийом таблеток.
Прогноз та прогноз
Прогноз при захворюванні на зберігання поганий. Причиною розладу здоров’я було виявлено генетичну диспозицію. Законодавчі вимоги забороняють медикам та вченим змінювати генетику людини. З цієї причини хвороба залишається на все життя і не має перспективи одужання.
Лікуючий лікар концентрується на лікуванні виниклих симптомів. Якщо їх не лікувати, з часом посилються різні скарги. Кісткова система пошкоджується і виникають проблеми органів. У гіршому випадку виникнуть функціональні труднощі у внутрішніх органах і в кінцевому рахунку збій діяльності органів. Це загрожує особі, яка стосується передчасної смерті.
Проблема захворювання полягає в діагностиці. У великої кількості пацієнтів помітні та сильно відчутні скарги виникають лише пізніше в житті. Як наслідок, генетичний розлад тривалий час залишається непоміченим, а раннє лікування захворювання утруднене. Чим пізніше поставлений діагноз, тим несприятливішим є подальший перебіг. На запущеній стадії захворювання внутрішні органи або суглоби вже сильно пошкоджені. Необхідні хірургічні втручання, і якщо хвороба прогресує несприятливо, лише один орган-донор може врятувати життя постраждалій людині. Тому раннє лікування є важливим для покращення прогнозу.
профілактика
Оскільки це вроджений генетичний дефект, який перешкоджає експресії ферменту, цю хворобу не можна лікувати профілактично. Однак останні досягнення в генній інженерії можуть забезпечити підхід у цій галузі.
Догляд за ними
При цьому захворюванні люди страждають від ряду різних ускладнень і недуг. Як правило, все це вкрай негативно впливає на якість життя постраждалої людини, тому діагноз слід ставити дуже рано. Чим раніше звертатися до лікаря, тим краще подальший перебіг цього захворювання.
Тяжкість цього захворювання може бути дуже різною, тому загальний прогноз часто неможливий. Постраждалі страждають від сильного ураження внутрішніх органів. У першу чергу страждають нирки і серце, тому дитина може померти в перші кілька днів, якщо симптоми не будуть вчасно усунені. Також є відкладення жиру в різних частинах тіла.
Особливо страждають пальці рук і ніг, що може призвести до значно зниженої естетики для постраждалої людини. Як правило, пошкодження нирок і головного мозку відбувається в подальшому перебігу, так що людина, яка постраждала, помирає внаслідок цього пошкодження. Батьки та родичі також часто страждають від депресії чи інших психічних розладів через хворобу.
Ви можете зробити це самостійно
Хвороби лізосомального зберігання дуже часто потребують інтенсивної медичної допомоги. Часто можливостей для самодопомоги недостатньо. Батьки постраждалих дітей часто відчувають сильний стрес в домашніх умовах, оскільки їх дитина потребує постійної турботи та уваги.
Клінічна картина окремих захворювань зберігання різна. Існують як легкі, так і дуже важкі форми. Одним із прикладів є хвороба Гоше. Допомога батьків часто обмежується годуванням дитини з обмеженими можливостями. У більш легких випадках тривалість життя може бути майже нормальною. Тим не менш, постійний нагляд лікаря необхідний для запобігання можливих ускладнень. Регулярні фізичні навантаження - одна з супутніх терапій, яку також можна проводити в домашніх умовах. Крім того, необхідно організувати ретельне обстеження на рак. Це вимагає постійних відвідувань лікаря з дитиною від батьків. Те ж стосується інших захворювань лізосомального зберігання.
У разі деяких захворювань, крім фізичних вад, можуть виникати і психічні обмеження, які ще потребують спеціальної підтримки. При легших формах деяких захворювань, таких як хвороба Хантера, спочатку трапляються лише зміни скелета та дисморфізм обличчя. Однак тут уражений хворий часто може вести самостійне життя. Однак тут необхідні постійні медичні огляди, щоб виключити можливі ускладнення, такі як серцева недостатність або респіраторні захворювання. Пацієнт може впоратися з психологічним стресом, викликаним фізичними деформаціями, через психологічні консультації.



.jpg)