The краніодіафізна дисплазія - це вроджене захворювання скелета, пов’язане з гіперостозом та склерозом лицьового черепа. Причиною є генетична мутація в генах, що гальмують кісткову структуру. Терапія є симптоматичною і фокусується на припиненні прогресування захворювання.
Що таке краніодіафізна дисплазія?

© crevis - stock.adobe.com
При гіперостозах кісткова речовина збільшується патологічно. Гіперостоз черепа - це група захворювань, пов’язана з таким збільшенням кісткової речовини в області черепа. Як краніодіафізна дисплазія характеризується вродженим гіперостозом черепа і є скелетним захворюванням.
Австралійський лікар Джон Халлідей вперше описав хворобу в середині 20 століття. Частота визначається з поширеністю менше одного випадку на 1 000 000 людей. Це робить захворювання скелета надзвичайно рідкісною дисплазією черепа.
Комплекс гіперостозу та стенозу лицьових та черепних кісток відстежується до генетичної причини. Через декілька зареєстрованих випадків до цього часу не всі зв’язки між хворобою були остаточно прояснені. З цієї причини на сьогоднішній день можливості терапії також обмежені.
причини
У великій кількості випадків краніодіафізна дисплазія виникає не епізодично, а при сімейному скупченні. Як аутосомно-рецесивний, так і аутосомно-домінантний спосіб успадкування були визначені як спосіб успадкування захворювання. Аутосомно-домінантна форма захворювання заснована на новій мутації гена SOST. Ген знаходиться в місці 17q21.31 і вважається одним з найважливіших інгібіторів утворення кісток.
Мутація генів SOST є причиною великої кількості спадкових захворювань кісток, таких як VDB. У разі мутації ген більше не може виконувати свої гальмівні функції, і кісткова структура розростається. Це принципово відрізняє гіперостоз краніодіафізної дисплазії від інших гіперостозів.
Більшість цих захворювань засновані на дисфункції остеокластів або остеобластів. Генетична диспозиція вважається доведеною у зв’язку із захворюванням. Які ще фактори відіграють роль у виникненні захворювання, остаточно не з’ясовано.
Симптоми, недуги та ознаки
Клінічна картина краніодіафізної дисплазії характеризується різними клінічними критеріями, які вже проявляються в грудному віці. У уражених немовлят зазвичай сильно закупорюються носові ходи, що може викликати у них проблеми з диханням. На пізньому перебігу захворювання в більшості випадків відбувається повна непрохідність носових ходів.
Часто слізні протоки пацієнта блокуються після цього явища. На нижній щелепі більшості уражених утворюються поступово наростаючі носові опуклі кісткові речовини. Гіперостоз лицьового черепа прогресує і переростає в леонтіазну осею. У більшості випадків розвиток зуба пацієнта порушується або затримується. Внутрішня частина черепа звужується в міру прогресування захворювання.
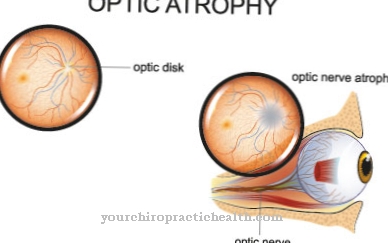
Перетяжки також впливають на фораміну і викликають послідовну атрофію зору. Це може супроводжуватися такими симптомами, як зниження слуху та більш-менш сильні головні болі. У деяких випадках, оскільки внутрішня частина черепа стає все більш вузькою, пацієнти також страждають від судом. Вали довгих трубчастих кісток розширюються все більше.
Діагностика та перебіг захворювання
Найбільш рання діагностика та подальша терапія значно покращують прогноз пацієнтів з краніодіафізною дисплазією. Лікар, мабуть, підозрює гіперостоз із візуального діагнозу. Процедури візуалізації вважаються найважливішим інструментом діагностики. Наприклад, рентген показує надзвичайний гіперостоз і склероз всіх кісток черепа.
Нашийники або ребра можуть здаватися розширеними в зображенні. Відсутня діафіза на довгих кістках чітко виділяється. Відмінна, не загущена кора також вписується в клінічну картину. З точки зору диференціальної діагностики, слід відрізняти такі захворювання, як синдром Енгельмана. Молекулярно-генетичні аналізи особливо підходять для такого диференційного діагнозу. Синдром Енгельмана показує зміни гена TGFB1 при мутаційному аналізі, тоді як краніодіафізна дисплазія впливає на ген SOST.
Ускладнення
Краніодіафізна дисплазія - рідкісна генетично обумовлена хвороба скелета. Симптом проявляється безпосередньо на лицьовому черепі через сильне збільшення кісткової речовини з супутнім склерозом. Генетична мутація проявляється вже в грудному віці за ознакою форми черепа та неправильно закладених носових ходів, що може спричинити загрозливі проблеми з диханням.
Отримані наслідки краніодіафізної дисплазії приносять ураженому пацієнту численні ускладнення, що обмежують життя з раннього віку. Якщо не буде своєчасного клінічного втручання, зростання надлишкової кістки буде прогресувати. Внутрішня частина черепа звужується, а рядки зубів формуються не адекватно. Потовщувальний кістковий матеріал звужує вушну раковину і є ризик порушення слуху і навіть зниження слуху.
У порожнині черепа зростає брак місця, а кісткові відкладення проникають у мозок. Виникають сильні головні болі, судоми, параліч обличчя та епілепсія, а також зменшення або погіршення розумово набутих навичок. Тому батьки, діти яких уражені краніодіафізною дисплазією, повинні шукати клінічних заходів на ранній стадії.
Після візуального обстеження диференціальна діагностика проводиться в межах заданих можливостей. В даний час немає базової терапії краніодіафізної дисплазії. Робляться спроби стримувати неконтрольоване прогресування росту кісток та його наслідки. Різні ліки, а також дієта зі зменшенням кальцію з самого немовляти допоможуть хворому зменшити симптоми.
Коли потрібно звертатися до лікаря?
Краніодіафізна дисплазія часто діагностується відразу після народження. Якщо це так, відповідальний лікар негайно повідомить батьків, а потім розпочне лікування безпосередньо. Якщо дисплазія менш виражена, діагноз ставлять батьки. Візит до лікаря показаний, якщо у новонародженого виникає утруднене дихання або водянисті очі. Зовнішні відхилення, такі як типові вади обличчя та зубів, також вказують на захворювання, яке потребує уточнення та лікування.
Батьки, які мають у дитини ознаки зниження слуху або судоми, повинні звернутися до лікаря. Це ж стосується, якщо дитина часто скаржиться на головні болі або відчуває сильний біль. Під час лікування дитину необхідно регулярно представляти до лікаря. Це забезпечить відновлення без ускладнень. Оскільки краніодіафізна дисплазія пов'язана з цілим рядом симптомів, терапія може зайняти місяці або навіть роки. З цією метою лікар загальної практики консультуватиметься з іншими фахівцями, завжди залежно від симптомів та скарг. Зазвичай до лікування залучаються неврологи, інтерністи, вушні фахівці, хірурги, фізіотерапевти та психологи.
Лікування та терапія
Причинно-наслідкової терапії для пацієнтів з краніодіафізною дисплазією ще не існує. Така терапія може бути можливою в майбутньому через підходи до генної терапії. Наразі, однак, захворювання можна лікувати лише симптоматично. Основна мета всіх терапевтичних заходів - зупинити надмірний ріст кісток. Існують різні кроки.
Прогресування хвороби можна, наприклад, зупинити за допомогою медикаментів. Кальцитріол і кальцитонін в основному використовуються як ліки. Оскільки структура кісток залежить від кальцію, дієта зі зменшенням кальцію також може мати сенс. Цю специфічну дієту слід застосовувати в довгостроковій перспективі і в ідеалі супроводжувати все життя пацієнта.
Медикаментозне лікування хворих на штучний глюкокортикоїдний преднізолон також показало позитивні ефекти. Чим раніше розпочата терапія, тим перспективніша перспектива. При надзвичайно ранньому лікуванні гіперостоз може бути припинений у перші роки життя. Таким чином різко зменшуються наступні симптоми.
За певних обставин хірургічні корекції також можуть бути внесені як частина терапії. Однак такі виправлення, як правило, мало сенсу, перш ніж перебіг хвороби буде підданий контролю.
Прогноз та прогноз
При вродженій, але дуже рідкісній краніодіафізній дисплазії спостерігається непоправна генетична мутація. Тому прогноз для постраждалих не дуже хороший. Медичні працівники можуть лише спробувати лікувати симптоми та наслідки посиленого росту кісток в області голови. Терапія може лише затримати перебіг захворювання. При краніодіафізній дисплазії збільшення кісткової речовини не зупиняється.
Оскільки сьогоднішні варіанти терапії не можуть змінити основну мутацію в стадії ембріона, від цього постраждають й інші покоління уражених. Сімейне скупчення помітно при краніодіафізній дисплазії. Симптоми, пов’язані з краніодіафізною дисплазією, вже можна помітити у немовляти. Оскільки всі спайки кісток відбуваються в області черепа, на них впливають верхні дихальні шляхи, а також слух або зір.
Крім того, на внутрішню частину черепа все більше впливає формування кісток. Це обмежує терапевтичні підходи для подальших скарг. Чим раніше можна поставити діагноз, тим краще прогноз довгостроковий. Дієта з низьким вмістом кальцію пригнічуватиме зростання росту кісток. Крім того, відповідні ліки та преднізолон можна вводити вже в грудному віці.
Міждисциплінарна стратегія лікування досягає найкращих результатів. Хірургічне втручання при краніодіафізній дисплазії має сенс лише за умови успішного стримування захворювання.
профілактика
Поки що немає профілактичних заходів щодо краніодіафізної дисплазії. Хвороба - це генетичне захворювання, яке пов’язане з сімейним диспозицією. Тому лише молекулярно-генетичне консультування може використовуватися як своєрідний профілактичний захід.
Догляд за ними
У більшості випадків у постраждалої людини доступно дуже мало заходів для подальшого спостереження. У деяких випадках це навіть може бути повністю обмеженим, так що людина, яка постраждала, залежить від чисто симптоматичного лікування захворювання. Самолікування не може відбутися, оскільки це генетичне захворювання.
Тому, якщо зацікавлена особа хоче народити дитину, їм слід провести генетичне обстеження та поради, щоб захворювання не повторилося у дітей. Само лікування зазвичай проводиться за допомогою різних препаратів, які можуть назавжди полегшити і обмежити симптоми. Завжди важливо стежити за тим, щоб його приймали регулярно, при цьому також слід дотримуватися правильної дозування.
Що стосується дітей, батьки, зокрема, повинні перевірити, чи правильно їх вживали та вживали. Регулярні огляди лікарем також необхідні для постійної перевірки стану хвороби. Більшість вад розвитку можна виправити за допомогою хірургічних втручань. Багато постраждалих також залежать від психологічної підтримки власної родини у повсякденному житті, що позитивно впливає на подальший перебіг захворювання. Як правило, це захворювання не скорочує тривалість життя пацієнта.
Ви можете зробити це самостійно
У разі краніодіафізної дисплазії уражений хворий має лише обмежені ефективні заходи, які позитивно впливають на перебіг захворювання. В першу чергу - це відповідна терапія краніодіафізної дисплазії командою фахівців. Хвороба починає проявляти себе в грудному віці, так що саме якість життя відповідних дітей сприяють насамперед батьки. У разі будь-якого стаціонарного перебування пацієнтів дитини, це часто має сенс, якщо батьки перебувають у лікарні, і дитина отримує в результаті емоційну підтримку.
В ході захворювання часто виникають порушення в розвитку зубів, тому пацієнти часто залежать від ортодонтичної терапії. Ваша власна співпраця також потрібна, коли мова йде про носіння брекетів. Також є дані, що дієта з низьким вмістом кальцію може стримувати прогресування краніодіафізної дисплазії. Тут також пацієнти мають значну свободу щодо їхньої співпраці, а отже, і якості життя.
Через проблеми з диханням пацієнти відмовляються від певних видів спорту, але також практикують зміцнювальні вправи вдома з фізіотерапевтом, якщо це дозволено медично. Діти з краніодіафізною дисплазією отримують адекватну освіту в спеціальних школах.
.jpg)



.jpg)
.jpg)


.jpg)