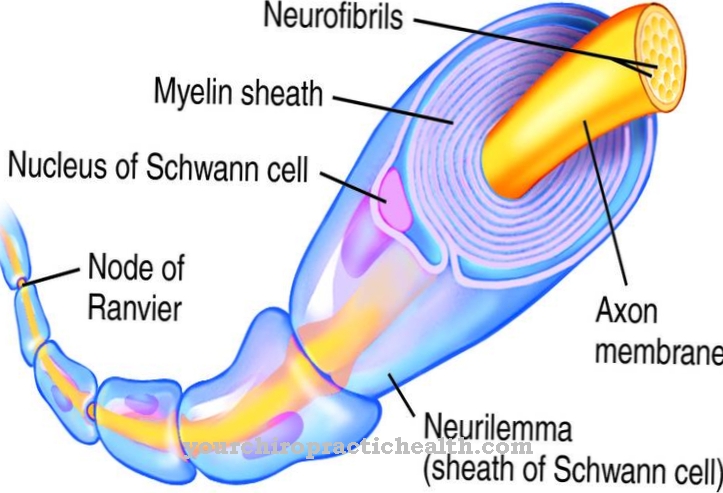
The Мукополісахаридоз є збірним терміном для захворювань зберігання лізосома, що ґрунтуються на зберіганні глікозаміногліканів. Усі захворювання розвиваються подібними симптомами і формами. Вираженість синдромів сильно різниться.
Що таке мукополісахаридоз?

© Себастьян Кауліцкі - stock.adobe.com
А Мукополісахаридоз не існує такого поняття, як окреме захворювання. Термін мукополісахаридоз - збірний термін для великої кількості захворювань зберігання, заснований на порушеннях зберігання глікозаміногліканів (GAG) у лізосомах клітин. Зберігання відбувається поступово, оскільки припинення з'єднань не працює.
Всі мукополісахаридози є генетичними. Кожному захворюванню не вистачає специфічного ферменту, який каталізує розпад відповідного GAG. Всі мукополісахаридози є дуже рідкісними захворюваннями і часто демонструють подібні курси. Якщо їх не лікувати, постійно зростаючі відкладення руйнують клітини. Органи руйнуються в процесі. Захворювання може початися в грудному віці, а також у дорослому віці.
Мукополісахаридоз може бути спричинений чотирма різними групами глікозаміногліканів:
- Гепарансульфат
- Кератан сульфат
- Хондроїтин сульфат
- Дерматан сульфат.
Всі глікозаміноглікани складаються з полісахаридного ланцюга, приєднаного до білка. Вуглеводний компонент складає 95 відсотків, а білковий компонент - п'ять відсотків молекулярної маси. Залежно від того, на який глікозаміноглікан та який фермент впливає, можна розрізняти шість основних основних форм мукополісахаридоз: До них відносяться хвороба Хурлера / Шея (MPS I), хвороба Хантера (MPS II), хвороба Санфіліппо (MPS III), хвороба Моркіо ( MPS IV), хвороба Марото-Ламі (MPS VI) та хвороба Слі (MPS VII). Всі типи мають важку і легку форму.
причини
Причиною всіх мукополісахаридоз є все більшого зберігання глікозаміногліканів (ГАГ) у лізосомах клітин. Деградація відповідних біополімерів порушена. При кожному окремому розладі відсутній або певний фермент, або цей фермент працює неправильно. Для кожного ферменту може бути декілька мутацій. Успадкування відповідної мутації може бути аутосомно-рецесивним, аутосомно-домінантним або рецесивним шляхом, пов'язаним з x.
Оскільки ферментативний процес, як правило, включає кілька етапів реакції, теоретично декілька ферментів можуть бути мутовані для одного і того ж глікозаміноглікану. Симптоми розладу були б однаковими чи подібними.
- В MPS I, Хвороба Гюрлера або Шеє, фермент альфа-l-ідуронідаза є дефектним.
- MPS II являє собою синдром мисливців з дефектною ідуронат-2-сульфатазою.
- Синдром Санфіліппо (MPS III) можна розділити на кілька підтипів. На цей стан можуть впливати кілька ферментів.
- Хвороба Моркіо (MPS IV) викликається дефектною β-галактозидазою.
- При синдромі Марото-Ламі (MPS VI) це N-ацетил-галактозамін-4-сульфат сульфатази.
- Хвороба Слі (MPS VII) викликається дефектною β-глюкуронідазою. Коли відповідні глікозаміноглікани зберігаються в лізосомах, вони стають все більшими та більшими.
Клітини також збільшуються, оскільки їм потрібно все більше місця для недеградованих ГАГ. Це також помітно при збільшенні багатьох органів. Типовим симптомом є постійне збільшення печінки та селезінки. Якщо їх не лікувати, хвороби зберігання призводять до смерті через поступове руйнування органів.
Симптоми, недуги та ознаки
Симптоми однакові для всіх захворювань. Існують важкі та легкі форми. Однак легкий перебіг лише означає, що хвороба прогресує повільніше. Підсумковий курс завжди однаковий. Спостерігається прогресуюча деформація скелетної системи, контрактури суглобів, огрубіння рис обличчя та збільшення печінки та селезінки.
Розумові та рухові навички знижуються в коротко- чи довгостроковій перспективі. При важких формах порушень клінічна картина дуже схожа. Пупкові та пахові грижі, проблеми з серцем та респіраторні інфекції виникають на ранній стадії. З часом звуження дихальних шляхів і розширення мигдалин і мигдалин переростають у масивні проблеми з апное.
Діагностика та перебіг захворювання
Мукополісахаридози можна діагностувати за допомогою дослідження сечі на глікозаміноглікани, що виділяються. При мукополісахаридозі значення завжди збільшуються. Також можна визначити активність підозрюваного дефектного ферменту в лейкоцитах або фібробластах. Певна схема виведення глікозаміногліканів призводить до підозри на відповідний фермент, який потім досліджують.
Ускладнення
Через мукополісахаридоз постраждалі страждають різними вадами розвитку та скелетними скаргами. Зустрічаються деформації, які можуть істотно обмежити повсякденне життя пацієнта. Як правило, суглоби також уражаються мукополісахаридозом, так що рух пацієнта обмежений.
Зокрема, діти страждають і страждають від сильно затримки розвитку, так що різні наслідкові ушкодження можуть траплятися і в дорослому віці. Не рідкість мукополісахаридоз може викликати проблеми в серці або диханні. У гіршому випадку раптова серцева смерть може призвести до смерті відповідної людини. Через утруднене дихання пацієнти страждають на втому та втому.
Стійкість постраждалих також сильно знижується. Не рідкість утруднене дихання вночі призводить до проблем зі сном і, отже, до депресії. Мукополісахаридоз значно знижує якість життя пацієнта. Причинно-наслідкове лікування цього захворювання, на жаль, неможливо. Тому постраждалі залежать від донорів кісткового мозку для лікування симптомів. Особливих ускладнень немає. Однак у більшості випадків пацієнти залежать від терапії протягом усього життя.
Коли потрібно звертатися до лікаря?
Зміни та відхилення в структурі тіла свідчать про порушення здоров'я. Візит до лікаря необхідний, як тільки виникають постійні оптичні особливості або у відповідної людини виникають труднощі свідомо оптимізувати свою поставу.Набряк суглобів, зміна рис обличчя або збільшення грудної клітки повинен інтенсивно обстежуватися у лікаря, щоб можна було поставити діагноз. Лікар потрібен, якщо є обмеження у можливостях руху, порушення в повсякденному добровільному контролі та зниження фізичної та розумової працездатності. Зверніться до лікаря у разі порушень серцевого ритму, проблем з диханням або перебоїв під час нічного сну.
Набряки в горлі, відчуття напруги в горлі, порушення в акті ковтання і зміни вокалізації вважаються тривожними. Їх повинен оглянути лікар, щоб симптоми могли бути полегшені. Якщо зацікавлена особа переносить більше інфекцій, якщо здатність сконцентруватися і звертати увагу падає або якщо пупкові або пахові грижі виникають повторно, слід повідомити лікаря про спостереження.
Необхідно обстежити та лікувати раптові вади, пожовтіння шкіри та внутрішній неспокій. Лікар потрібен, як тільки з’являються болі в тілі, знижена якість життя та проблеми з поведінкою. Якщо є ризик задухи, потрібна швидка допомога. Для запобігання цього гострого стану слід якомога раніше звернутися до лікаря.
Терапія та лікування
Причинно-наслідкова терапія сьогодні ще не можлива. Однак у дослідницьких проектах є деякі підходи до майбутньої генної терапії цих захворювань. На жаль, на даний момент відчутних результатів у цій галузі немає. Однак клінічне дослідження генотерапії хвороби Гурлера має розпочатися в Барселоні. При деяких формах мукополісахаридозів перенесення кісткового мозку виявився ефективним в окремих випадках. Це впливає, наприклад, на хворобу Хантера, хворобу Гурлера або хворобу Санфіліппо.
За допомогою цього перенесення кісткового мозку хворі стовбурові клітини обмінюються на здорові стовбурові клітини від донора. Це дає можливість організму достатньо відновити відсутні фермент. Ензимозамісна терапія також окупається у багатьох випадках. Однак цю замісну терапію необхідно проводити довічно. Однак є й випадки, коли перспективні методи терапії вже неможливі. Однак суть тут полягає в проведенні симптоматичних методів лікування.
Ви можете знайти свої ліки тут
➔ Ліки від болюПрогноз та прогноз
Подальший розвиток у пацієнтів із мукополісахаридозом необхідно оцінювати індивідуально. Цей термін є збірним терміном для різних захворювань зберігання. Вони є різними ступенями у кожного пацієнта і їх інтенсивність виражена індивідуально. Якщо медична допомога не буде розпочата, внутрішні органи всіх постраждалих поступово руйнуються протягом життя. Це призводить до скорочення середнього очікуваного життя.
При ранньому діагнозі може бути розроблена персонально оптимізована терапія. Це пов'язано з вимогами до здоров'я та наявними скаргами пацієнта. Тривале лікування принципово необхідне для досягнення стабільного покращення здоров’я. Можуть відбуватися хірургічні втручання, кожне з яких пов’язане з різними ризиками та побічними ефектами. Якщо операція протікає без будь-яких подальших ускладнень, симптоми зазвичай полегшуються після цього.
Тим не менш, небажані події та невдачі можуть виникнути протягом життя. В окремих випадках лише трансплантація кісткового мозку може покращити загальну якість життя. Через загальні обставини пацієнт відчуває сильне емоційне та психічне навантаження. Нормальне повсякденне життя часто неможливо через симптоми. Психологічні ускладнення можуть виникнути і призвести до подальшого погіршення ситуації.
профілактика
Оскільки мукополісахаридози є спадковими захворюваннями, профілактика неможлива. У разі наявного захворювання успіх лікування можна забезпечити шляхом своєчасної терапії. Крім того, необхідний постійний моніторинг роботи легенів та серця. Якщо випадки мукополісахаридозу вже відбулися в сім’ї, ризик захворювання можна оцінити за допомогою генетичного консультування, якщо сім'я бажає мати дітей.
Догляд за ними
У більшості випадків мукополісахаридозу у пацієнта є лише кілька варіантів подальшої допомоги, так що особа, яка постраждала, повинна в першу чергу проконсультуватися з лікарем на ранній стадії. Тільки при ранньому виявленні та лікуванні цього захворювання можна запобігти подальшим ускладненням, тому слід негайно звернутися до лікаря, як тільки з’являться перші ознаки та симптоми.
У більшості випадків постраждалі залежать від хірургічних втручань, які можуть полегшити та обмежити симптоми. Однак, оскільки мукополісахаридоз є генетичним захворюванням, його зазвичай неможливо повністю вилікувати.
Тому зацікавлена особа повинна спочатку звернутися до лікаря, якщо вони хочуть мати дітей, щоб запобігти рецидиву цього захворювання у дітей. Часто також дуже важливо мати підтримку сім'ї під час лікування. Це також може запобігти депресію та інші психологічні розлади. Мукополісахаридоз може призвести до скорочення тривалості життя постраждалої людини, внаслідок чого подальший перебіг дуже залежить від часу постановки діагнозу.
Ви можете зробити це самостійно
Можливості самодопомоги при мукополісахаридозі обмежуються полегшенням симптомів і тим самим покращує якість життя. Групи самодопомоги виявилися дуже корисними, оскільки обмін з іншими батьками виявляє цінні поради та часто може полегшити страхи та занепокоєння та дати більш позитивний погляд на майбутнє.
Супровід фізіотерапії, трудотерапії, логопедії та інших форм терапії, які часто можна поглибити в домашніх умовах, зараз є невід’ємною частиною життя.
Для того, щоб зробити життя максимально легким для себе та постраждалої дитини, доцільно зробити житлове середовище доступним для інвалідів якомога раніше. Зі збільшенням віку та ваги дитини ліжка для догляду за регулюванням висоти виявляються прекрасним фізичним полегшенням для вихователя. Пристрої попередження епілепсії та інші технічні засоби дозволяють забезпечити найкращу безпеку навіть вночі та полегшити батьків вночі, щоб вони могли спати спокійніше.
Ведення щоденника симптомів може допомогти лікареві розпізнати нові симптоми та, можливо, скорегувати лікування існуючих симптомів, оскільки медикаментозна терапія часто виявляє не бажаний ефект, а протилежний ефект.
Оскільки хвороба дуже вимоглива до родичів, їм доводиться створювати мало місця для підзарядки акумуляторів. Сюди можна віднести лікування, профілактичну допомогу або пізніше відпустку в хоспісі.

.jpg)



.jpg)
.jpg)


.jpg)