The Ензимна замісна терапія використовується для лікування захворювань лізосомального зберігання, при яких нестача ферментів призводить до патологічного накопичення продуктів розпаду в лізосомах клітин.
Відсутні ферменти через генетичні дефекти компенсуються регулярними внутрішньовенними вливаннями. Оскільки влиті синтетичні ферменти не можуть перетинати гематоенцефалічний бар'єр через їх молекулярний розмір, терапія працює лише при захворюваннях лізосомального зберігання, які не впливають на центральну нервову систему.
Що таке ензимна замісна терапія?

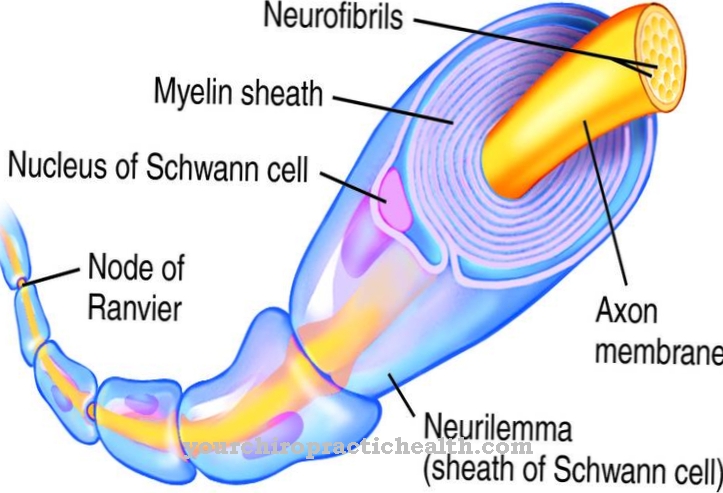
Лізосоми - це спеціальні клітинні органели, в яких чужорідні та ендогенні речовини розщеплюються та частково переробляються. Для деградації та транспортування речовин необхідні специфічні гідролізуючі ферменти. Це протеази, нуклеази, ліпази та транспортерні речовини.
Ряд відомих генетичних дефектів може призвести до виходу з ладу певних ферментів, так що деякі продукти деструкції накопичуються в лізосомах у патологічних кількостях і накопичуються до тих пір, поки вони не надходять на позаклітинний матрикс, тобто міжклітинні простори. Усі генетичні дефекти, що призводять до збою хоча б однієї необхідної гідролази, підсумовуються під терміном хвороба лізосомального зберігання. Ензимозамісна терапія (ERT, ензимна замісна терапія) використовується для заміни відсутніх ендогенних ферментів синтетично виробленими ферментами.
Оскільки гідролази складаються з відносно великих молекул, вони не можуть бути поглинені з кишечника, попередньо не розбившись та інактивуючись, так що їх можна вводити лише за допомогою внутрішньовенної інфузії. Однак розмір молекул ферменту також перешкоджає перетину гематоенцефалічного бар'єру, так що терапія може бути ефективною лише при захворюваннях зберігання лізосом, які не впливають на центральну нервову систему (ЦНС).
Функція, ефект та цілі
Відомо понад 50 різних лізосомальних метаболічних порушень, кожне з яких простежується до моногенетичного дефекту. Хвороби лізосомального зберігання можна розділити на сім різних класів залежно від надмірно зберігаються речовин через наявний ферментний дефект.
Мукополісахаридози та олігосахаридози в першу чергу підходять для ЕРТ. Метою ERT завжди є компенсувати дефіцит специфічного ферменту за допомогою штучно поставлених ферментів, щоб привести хворобу в глухий кут або принаймні більш м'який перебіг. Детально доступні замісні ферменти для наступних захворювань лізосомального зберігання:
- Хвороба Гоше
- Хвороба Помпе
- Хвороба Фабрі
- Синдром Герлера-Пфаундлера (мукополісахаридоз I)
- Хвороба мисливця (мукополісахаридоз II)
• синдром Марото-Ламі (мукополісахаридоз VI) • Німан-Пік Б
Хвороба Гоше - найпоширеніша хвороба лізосомального зберігання. Він зустрічається у трьох різних варіантах, два з яких також впливають на нервову систему. При ненейропатичній формі особливо страждає селезінка, що значно збільшується і призводить до вторинних пошкоджень, таких як анемія та пошкодження кісткового мозку. Типовими симптомами є біль у кістках та суглобах та порушення кровообігу. Гострий нейропатичний варіант захворювання демонструє важкий перебіг і надає мало шансів на виживання після перших двох років життя.
Хвороба Помпе зберігання зумовлена дефіцитом ферменту альфа-1,4-глюкозидази, який бере участь у великій кількості обмінних процесів. Хвороба Помпе призводить до величезного збільшення серця (кардіомегалія) та серцевої недостатності. Існують ранні, серйозні курси, які з’являються в перші кілька місяців життя, а також легші форми, які з’являються лише в пізніші роки життя.
Хвороба Фабрі викликається генетичним дефектом, пов'язаним з Х, тому хворобою зберігання можуть бути уражені лише хлопчики та чоловіки. Захворювання зазвичай призводить до симптомів у похилому віці, включаючи напади болю, кератоми шкіри, проблеми з нирками та ураження серцевих м’язів. Дефіцит ферменту альфа-галактозидази А призводить до накопичення церамідного тригексозиду, що є причиною запуску симптомів, що також може впливати на вегетативну нервову систему.
Не рідкість пошкодження може призвести до інфаркту, інфаркту нирки або навіть до інсульту. Синдром Гурлера-Пфаундлера також відомий як мукополісахаридоз I типу і викликаний порушенням метаболізму глікозаміногліканів. Захворювання пов’язане з найрізноманітнішими симптомами, включаючи важкі порушення психіки та серйозні зміни скелета. Перебіг захворювання важкий, так що середня тривалість життя задається як 11 - 14 років. Хвороба Хантера відповідає мукополісахаридозу 2 типу і, як хвороба Герлера, - викликана дефектом, пов'язаним з Х. Хвороба характеризується курсами різної тяжкості, від виникнення в ранньому дитячому віці до легких курсів, які з’являються лише у дорослих чоловіків.
Через найпоширеніших серцевих симптомів, таких як вади серцевого клапана та проблеми із серцевим м’язом, тривалість життя коливається від нормальної до незначно обмеженої. Синдром Марото-Ламі (MPS VI) є однією з мукополісахаридоз, які успадковуються автосомно-рецесивно, оскільки дефект гена збудника не знаходиться на Х-хромосомі. Захворювання зустрічається дуже рідко, зафіксовано один випадок на 455 000 народжених. Відомі легкі та важкі форми.
Симптомами є збільшена печінка та селезінка, синдром зап’ястного каналу та зміни серцевих клапанів. Niemann-Pick B - це сфінгомієліновий ліпідоз, який є одним із захворювань лізосомального зберігання і викликаний генетичним дефектом на хромосомі 11. Хоча тип B захворювання вражає переважно печінку та селезінку, тип А також має значні нейронні проблеми.
Ви можете знайти свої ліки тут
➔ Ліки від болюРизики, побічні ефекти та небезпеки
Оскільки багато захворювань лізосомального зберігання, які можна лікувати за допомогою ензимозамісної терапії, приймають важкий перебіг із відповідно збільшеним показником смертності, найбільший ризик ERT полягає в тому, що вибраний замісний фермент не працює або працює надто слабко.
Інший ризик полягає в меншій мірі самої терапії, ніж у тому, що основне захворювання розпізнається занадто пізно, так що ЕРТ може припинитися протягом курсу, але вже заподіяний збиток не може знову регресувати. Приблизно кожен другий лікуваний пацієнт тимчасово реагує на інфузії з такими симптомами, як лихоманка та озноб. Причини цього ще не повністю з'ясовані. Деякі пацієнти реагують, утворюючи антитіла, і були відомі випадки, коли пацієнти реагували на висип і бронхоспазм.


.jpg)



.jpg)
.jpg)


.jpg)