Лізил гідроксилази являють собою групу ферментів, які відповідають за гідроксилювання залишків лізину всередині білків. Так вони в основному сприяють структурі сполучної тканини. Порушення функції лізил гідроксилази виражаються при таких захворюваннях, як цинга або спадковий синдром Елерса-Данлоса.
Що таке лізил гідроксилази?
Лізильні гідроксилази - це ферменти, завданням яких є каталізація посттрансляційної модифікації амінокислоти лізин шляхом включення гідроксильної групи в гідроксильний лізин. Це зміцнює сполучну тканину, оскільки її білкові ланцюги надають можливість подальшої мережі через гідроксильні групи.
Люзил гідроксилаза людини складається з 727 амінокислот. Лізильні гідроксилази також належать до групи гідроксилаз, тобто ферментів, які загалом каталізують включення гідроксильних груп у молекули. Крім лізил гідроксилаз, до складу гідроксилаз або оксидоредуктаз входять також проліл гідроксилази, фенілаланін гідроксилаза, тирозин гідроксилаза або триптофан гідроксилаза. Особливо разом з пролілгідроксилазами лізил гідроксилази відіграють важливу роль у функції сполучної тканини. Обидві групи ферментів потребують коензиму вітамін С для своєї функції.
Функція, ефект та завдання
Функція лізилгідроксилаз складається виключно з каталізації включення гідроксильних груп у залишки лізину всередині білка. В ході посттрансляційної модифікації амінокислота гідроксиламін утворюється з лізину.
Хоча гідроксиламін також вільний, він не може бути включений до білка в цій формі. Отже, посттрансляційна модифікація означає подальшу конверсію цієї амінокислоти після накопичення білка. Коли атом водню обмінюється на гідроксильну групу, в цей бік вбудована функціональна група, яка може виконувати мостикові функції. За допомогою гідроксильної групи різні білкові ланцюги можуть зв'язуватися між собою. Крім того, молекули цукру можуть зв'язуватися з цією функціональною групою. Обидві реакції дуже важливі, серед іншого, у розвитку сполучної тканини.
Сполучна тканина охоплює організм і внутрішні органи. Він повинен бути твердим і підтягнутим, щоб можна було диференціювати функціонально різні органи. Це забезпечується білками сполучної тканини, які містять високий відсоток амінокислот лізину та проліну.Для цього обидві амінокислоти згодом частково модифікуються після їх включення в білок шляхом додавання гідроксильної групи. Як вже було сказано, при проліні ця реакція каталізується проліл гідроксилазами, а лізин - лізилгідроксилазами. Після утворення білка ці реакції модифікації створюють мережу білкових ланцюгів, які представляють собою щільну сполучну тканину.
Без функції обох ферментів розвиток функціональної сполучної тканини був би взагалі неможливим. Однак обидва ферменти працюють лише за допомогою коферменту аскорбінової кислоти, тобто вітаміну С. При структурно змінених ферментах через мутацію або нестачі вітаміну С це може призвести до порушень у структурі сполучної тканини і, отже, до серйозних захворювань.
Освіта, виникнення, властивості та оптимальні значення
Ген PLOD1 відповідає за кодування людської лізил гідроксилази. Назва PLOD1 походить від назви лізилгідроксилази "проколаген лізин, 2-оксоглутарат-5-діоксигеназа 1". Цей ген розташований на хромосомі 1. Оскільки постійно утворюється нова сполучна тканина, існує також постійна потреба у виробництві лізилгідроксилаз. Тому мутація цього гена може мати дуже серйозні наслідки для здоров’я організму.
Хвороби та розлади
Порушення функції лізил гідроксилази відіграють особливо важливу роль при цинзі та синдромі Елерса-Данлоса. Шкурка відома як давня хвороба моряків, яка викликана нестачею вітаміну С. Вітамін С, також відомий як аскорбінова кислота, функціонує як коензим лізил гідроксилази та проліл гідроксилази. Якщо його не вистачає, амінокислоти лізин та пролін у білку сполучної тканини вже не можуть гідроксилюватися.
Оскільки відбувається постійне накопичення та розпад білків сполучної тканини, протеїнові ланцюги все менше і менше здатні до мережі в період дефіциту вітамінів. Сполучна тканина слабшає і більше не може належним чином виконувати свою функцію. Зустрічаються найрізноманітніші симптоми, включаючи загальне виснаження, сприйнятливість до інфекції, кровоточивість ясен, втрата зуба, погане загоєння ран, серйозні проблеми зі шкірою, втрата м’язів та багато інших порушень здоров’я. Шкірка може в кінцевому рахунку призвести до смерті від загальної серцевої недостатності або важких інфекцій. Стародавні мореплавці особливо постраждали, оскільки вони не могли отримувати достатню кількість вітаміну С під час тривалих плавань у морі.
Було показано, що хвороба заживає негайно, коли даються певні продукти, такі як квашена капуста. Лише пізніше було визнано, що причиною захворювання був дефіцит вітаміну С. Спалах хвороби цинги моряка згодом був запобіжений годуванням моряків квашеною капустою. Ще одне захворювання, яке лише частково можна віднести до дефекту лізилгідроксилази людини, - синдром Елера-Данлоса. Синдром Елера-Данлоса - збірний термін для різних спадкових захворювань сполучної тканини з різними причинами. Цей синдром характеризується вираженою слабкістю сполучної тканини.
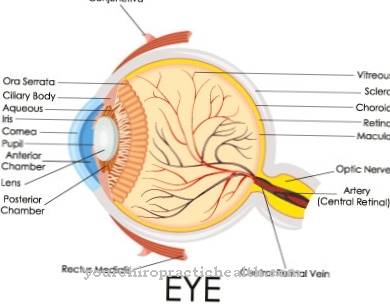
Шкіра перетягується, а суглоби - нерухомі. Генетично модифікована лізил гідроксилаза запускає синдром Елера-Данлоса типу VI. Мутаційний ген під назвою PLOD1, який знаходиться на хромосомі 1, відповідає за це. Утворений з цього дефектний фермент вже не повністю функціонує і може лише недостатньо каталізувати реакції гідроксилювання на лізин. Розвивається слабка сполучна тканина з відомими симптомами, а також додаткове ураження очей та внутрішніх органів. Синдром Елерса-Данлоса типу VI може успадковуватися як аутосомно-рецесивна ознака.



.jpg)
.jpg)




-eisenmangelanmie.jpg)